Using multiple stains on Xenium data#
%load_ext autoreload
%autoreload 2
%matplotlib inline
import json
import os
from pathlib import Path
import pandas as pd
import pooch
from spatialdata import SpatialData, read_zarr
from spatialdata.models import TableModel
from spatialdata.transformations import Scale, Sequence, get_transformation, set_transformation
from spatialdata_io import xenium_aligned_image
from spatialdata_io._constants._constants import XeniumKeys
import matplotlib.pyplot as plt
import spatialdata as sd
import numpy as np
import tempfile
import sparrow as sp
from sparrow.datasets.registry import get_registry
Read in data#
# If path is set to None, example data will be downloaded in the default cache folder of your os. Set this to a custom path to change this behaviour.
path = None
registry = get_registry( path = path ) # on Windows, set path (e.g. to r"c:\tmp")
path_unzipped = registry.fetch(
"transcriptomics/xenium/Xenium_V1_humanLung_Cancer_FFPE/Xenium_V1_humanLung_Cancer_FFPE_outs.zip", # Xenium output bundle
processor=pooch.Unzip(extract_dir="."),
)
_ = registry.fetch(
"transcriptomics/xenium/Xenium_V1_humanLung_Cancer_FFPE/Xenium_V1_humanLung_Cancer_FFPE_he_image.ome.tif" # post-Xenium H&E stain
)
_ = registry.fetch(
"transcriptomics/xenium/Xenium_V1_humanLung_Cancer_FFPE/Xenium_V1_humanLung_Cancer_FFPE_he_imagealignment.csv" # H&E image alignment
)
input_path = os.path.commonpath(path_unzipped)
# The OUTPUT_DIR is the directory where the SpatialData .zarr will be saved. Change it to your output directory of choice.
OUTPUT_DIR = tempfile.gettempdir()
# Read in Xenium data using SPArrOW's Xenium reader.
sdata = sp.io.xenium(
input_path,
to_coordinate_system="global",
aligned_images=True,
cells_table=False,
nucleus_labels=True,
cells_labels=True,
filter_gene_names=['Unassigned','NegControl'],
# output= os.path.join(OUTPUT_DIR, f"sdata_{uuid.uuid4()}.zarr"),
output= os.path.join(OUTPUT_DIR, f"sdata_Xenium_lung_multistaining_tutorial.zarr")
)
sdata
SpatialData object, with associated Zarr store: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr
├── Images
│ ├── 'he_image_global': DataTree[cyx] (3, 45087, 11580), (3, 22543, 5790), (3, 11271, 2895), (3, 5635, 1447), (3, 2817, 723)
│ └── 'morphology_focus_global': DataTree[cyx] (4, 17098, 51187), (4, 8549, 25593), (4, 4274, 12796), (4, 2137, 6398), (4, 1068, 3199)
├── Labels
│ ├── 'cell_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
│ └── 'nucleus_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
└── Points
└── 'transcripts_global': DataFrame with shape: (<Delayed>, 3) (2D points)
with coordinate systems:
▸ 'global', with elements:
he_image_global (Images), morphology_focus_global (Images), cell_labels_global (Labels), nucleus_labels_global (Labels), transcripts_global (Points)
# For some reason, the interior RNA (S18) and boundary (ATP1A1 / CD45 / E-Cadherin) channel names are switched here, we will switch them back
sdata['morphology_focus_global']["scale0"]["image"].c.data
img = sdata['morphology_focus_global']
for i in range(4):
scale = f"scale{i}"
old_names = list(img[scale].coords["c"].values)
old_names[1], old_names[2] = old_names[2], old_names[1]
img[scale].coords["c"] = old_names
img[scale].coords["c"]
sdata['morphology_focus_global'] = img
sdata['morphology_focus_global']["scale0"]["image"].c.data
array(['DAPI', '18S', 'ATP1A1/CD45/E-Cadherin', 'AlphaSMA/Vimentin'],
dtype='<U22')
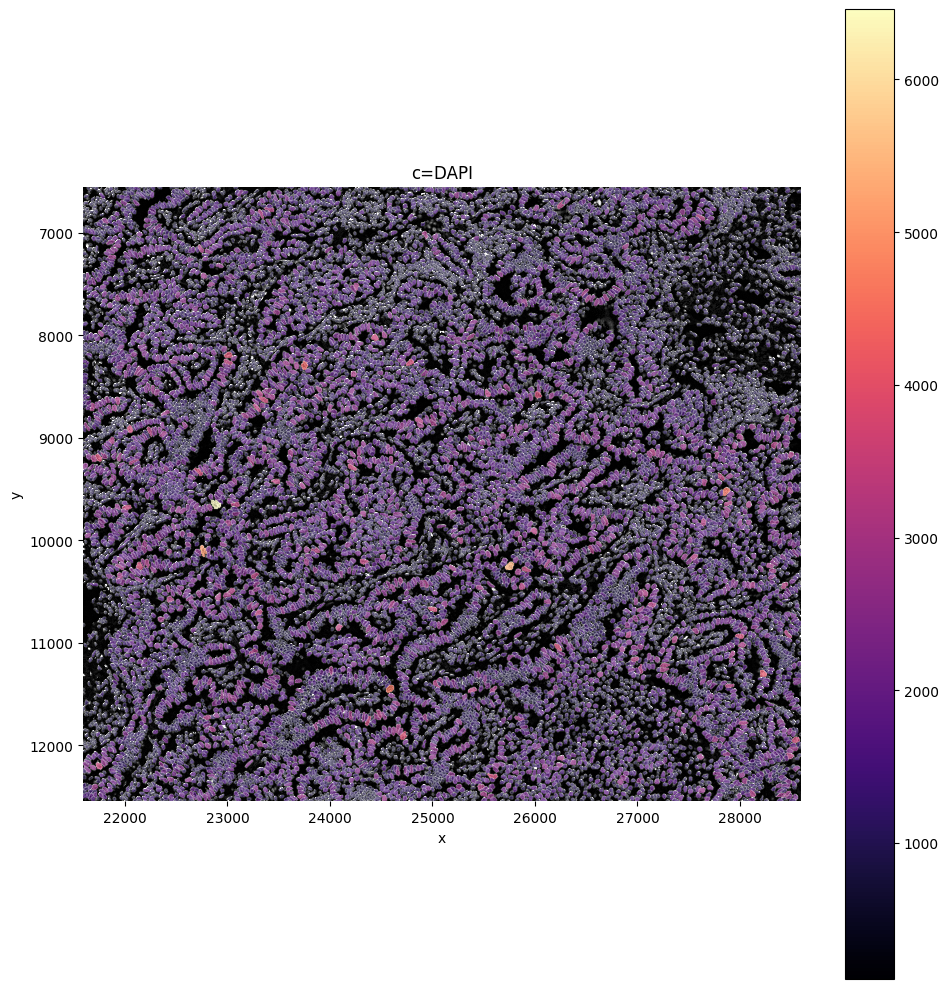
# Check with crop if channels are as expected
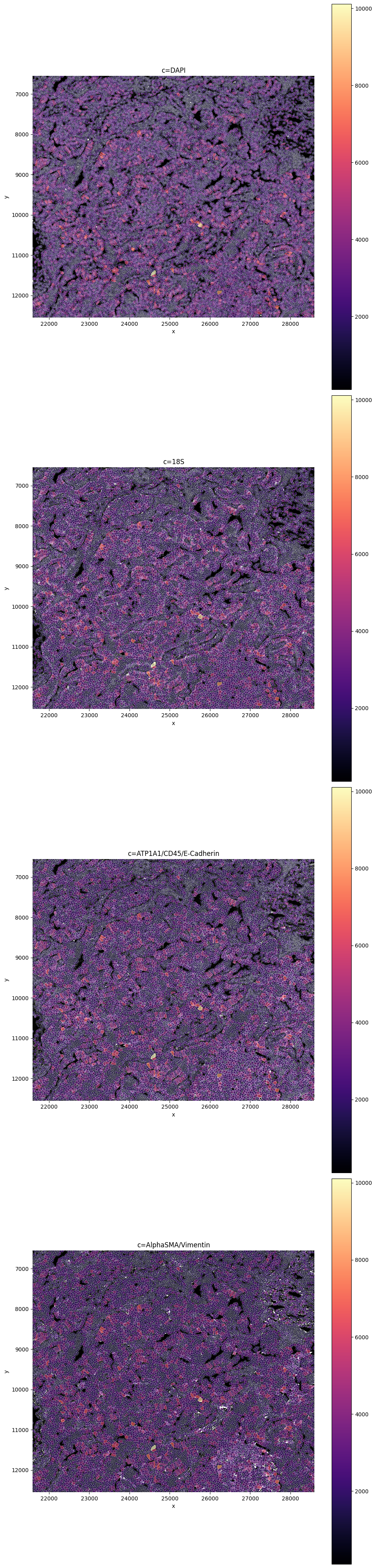
crd = [21593, 28593, 6549, 12549]
sp.pl.plot_image(sdata, img_layer="morphology_focus_global", crd=crd, to_coordinate_system='global')

In this tutorial, we demonstrate three different ways to use SPArrOW to segment cells in a multi-modal image. First, we perform segmentation using only the DAPI channel, which yields nuclear information only. Next, we show how to combine segmentations from the DAPI image, the ATP1A1 / CD45 / E-Cadherin (boundary stain) image, and the S18 (interior RNA stain) image, following an approach comparable to the default 10X Xenium analysis pipeline. Finally, we demonstrate how to jointly use the nuclei and boundary channels as input to the Cellpose segmentation model.
We will first preprocess the image to make it as clean as possible for the segmentation. For more details on these steps, we refer to the general “SPArrOW: How to start” tutorial.
sdata = sp.im.min_max_filtering(
sdata,
img_layer="morphology_focus_global",
output_layer="min_max_filtered",
size_min_max_filter=[55, 185, 60, 45], # specify a single value to be used for all channels, or specify a list with a value for each channel
crd = [21593, 28593, 6549, 12549],
overwrite=True,
)
sdata = sp.im.enhance_contrast(
sdata,
img_layer="min_max_filtered",
output_layer="clahe",
contrast_clip=[25, 1, 15, 2], # specify a single value to be used for all channels, or specify a list with a value for each channel
overwrite=True,
)
# Display the result of preprocessing the staining image channels.
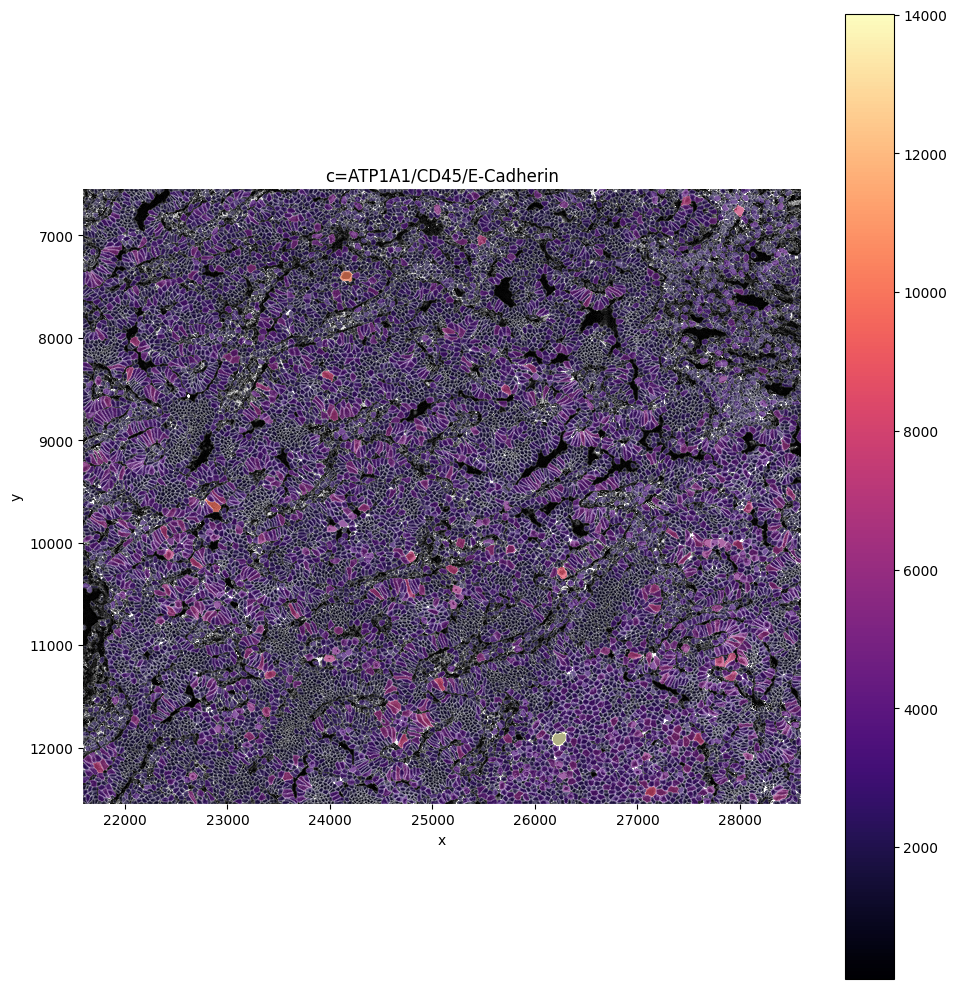
sp.pl.plot_image(
sdata,
img_layer=["morphology_focus_global", "clahe"],
channel=['DAPI', '18S', 'ATP1A1/CD45/E-Cadherin'],
crd=[21593, 28593, 6549, 12549],
)
# Orignal image is plotted on the left, preprocessed image on the right.

Cell segmentation: DAPI (only nuclei)#
# Separate the DAPI channel
sdata["DAPI"] = sdata["clahe"][0:1, :, :]
sdata
SpatialData object, with associated Zarr store: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr
├── Images
│ ├── 'DAPI': DataArray[cyx] (1, 6000, 7000)
│ ├── 'clahe': DataArray[cyx] (4, 6000, 7000)
│ ├── 'he_image_global': DataTree[cyx] (3, 45087, 11580), (3, 22543, 5790), (3, 11271, 2895), (3, 5635, 1447), (3, 2817, 723)
│ ├── 'min_max_filtered': DataArray[cyx] (4, 6000, 7000)
│ └── 'morphology_focus_global': DataTree[cyx] (4, 17098, 51187), (4, 8549, 25593), (4, 4274, 12796), (4, 2137, 6398), (4, 1068, 3199)
├── Labels
│ ├── 'cell_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
│ └── 'nucleus_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
└── Points
└── 'transcripts_global': DataFrame with shape: (<Delayed>, 3) (2D points)
with coordinate systems:
▸ 'global', with elements:
DAPI (Images), clahe (Images), he_image_global (Images), min_max_filtered (Images), morphology_focus_global (Images), cell_labels_global (Labels), nucleus_labels_global (Labels), transcripts_global (Points)
with the following Dask-backed elements not being self-contained:
▸ DAPI: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
with the following elements not in the Zarr store:
▸ DAPI (Images)
# Segment the cells, we advise to optimize the hyperparameters on a crop, to make the process quicker
crd=[21593, 28593, 6549, 12549]
sdata = sp.im.segment(sdata=sdata,
img_layer='DAPI',
output_labels_layer='segmentation_mask_dapi',
output_shapes_layer='segmentation_mask_boundaries_dapi',
crd=crd,
to_coordinate_system='global',
flow_threshold=0.90,
diameter=25,
cellprob_threshold=-4,
min_size=15,
pretrained_model="nuclei",
overwrite=True)
sp.pl.segment(sdata=sdata, crd=crd, img_layer="DAPI", shapes_layer="segmentation_mask_boundaries_dapi")

# Allocation of the transcripts to the segmentation mask
sdata = sp.tb.allocate(
sdata=sdata,
labels_layer="segmentation_mask_dapi",
points_layer="transcripts_global",
output_layer="table_dapi",
update_shapes_layers=False,
overwrite=True,
)
sdata['table_dapi']
AnnData object with n_obs × n_vars = 19894 × 377
obs: 'cell_ID', 'fov_labels'
uns: 'spatialdata_attrs'
obsm: 'spatial'
# Perform preprocessing
sdata = sp.tb.preprocess_transcriptomics(
sdata,
labels_layer="segmentation_mask_dapi",
table_layer="table_dapi",
output_layer="table_dapi_preprocessed",
min_counts=10,
min_cells=5,
size_norm=True,
n_comps=50,
overwrite=True,
update_shapes_layers=True,
shapes_layers_to_filter=['segmentation_mask_boundaries_dapi']
)
# NOTE: We're skipping a bunch of QC-steps here. For more details about this, see general tutorial
# we can plot cell sizes
sp.pl.plot_shapes(
sdata,
img_layer="DAPI",
table_layer="table_dapi_preprocessed",
column="shapeSize",
crd=crd,
shapes_layer="segmentation_mask_boundaries_dapi",
linewidth=0.2,
alpha=0.7
)
# We can further filter on cell size in order to discard any segmentation mistakes
sdata = sp.tb.filter_on_size(
sdata,
labels_layer="segmentation_mask_dapi",
table_layer="table_dapi_preprocessed",
output_layer="table_dapi_preprocessed",
min_size=150,
max_size=5000,
overwrite=True,
shapes_layers_to_filter=['segmentation_mask_boundaries_dapi']
)
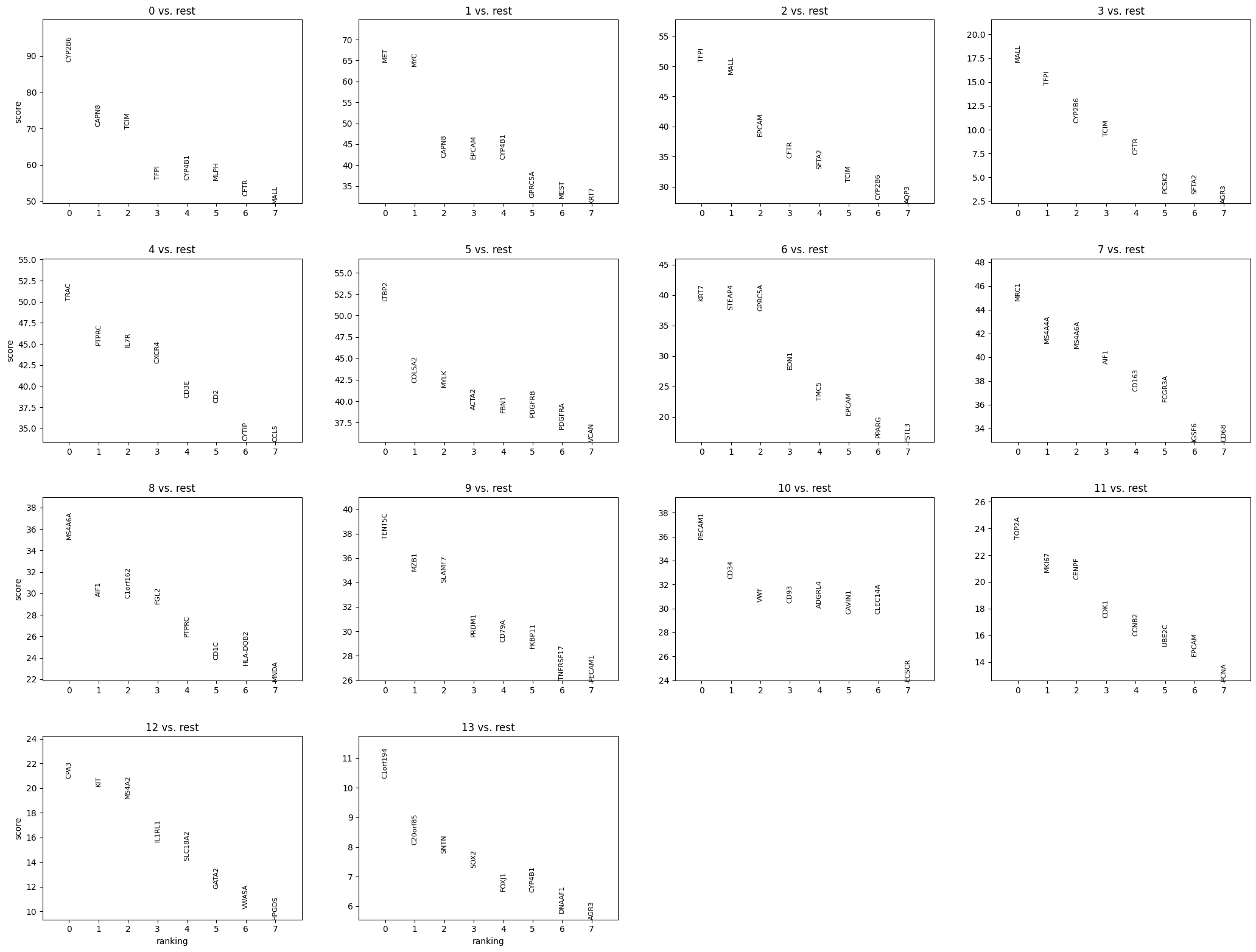
# Now we can cluster our data
sdata = sp.tb.leiden(
sdata,
labels_layer="segmentation_mask_dapi",
table_layer="table_dapi_preprocessed",
output_layer="table_dapi_preprocessed",
calculate_umap=True,
calculate_neighbors=True,
n_pcs=17,
n_neighbors=35,
resolution=0.8,
rank_genes=True,
key_added="leiden",
overwrite=True,
)
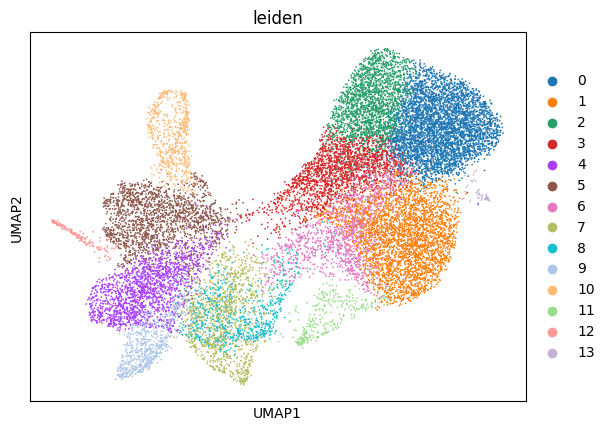
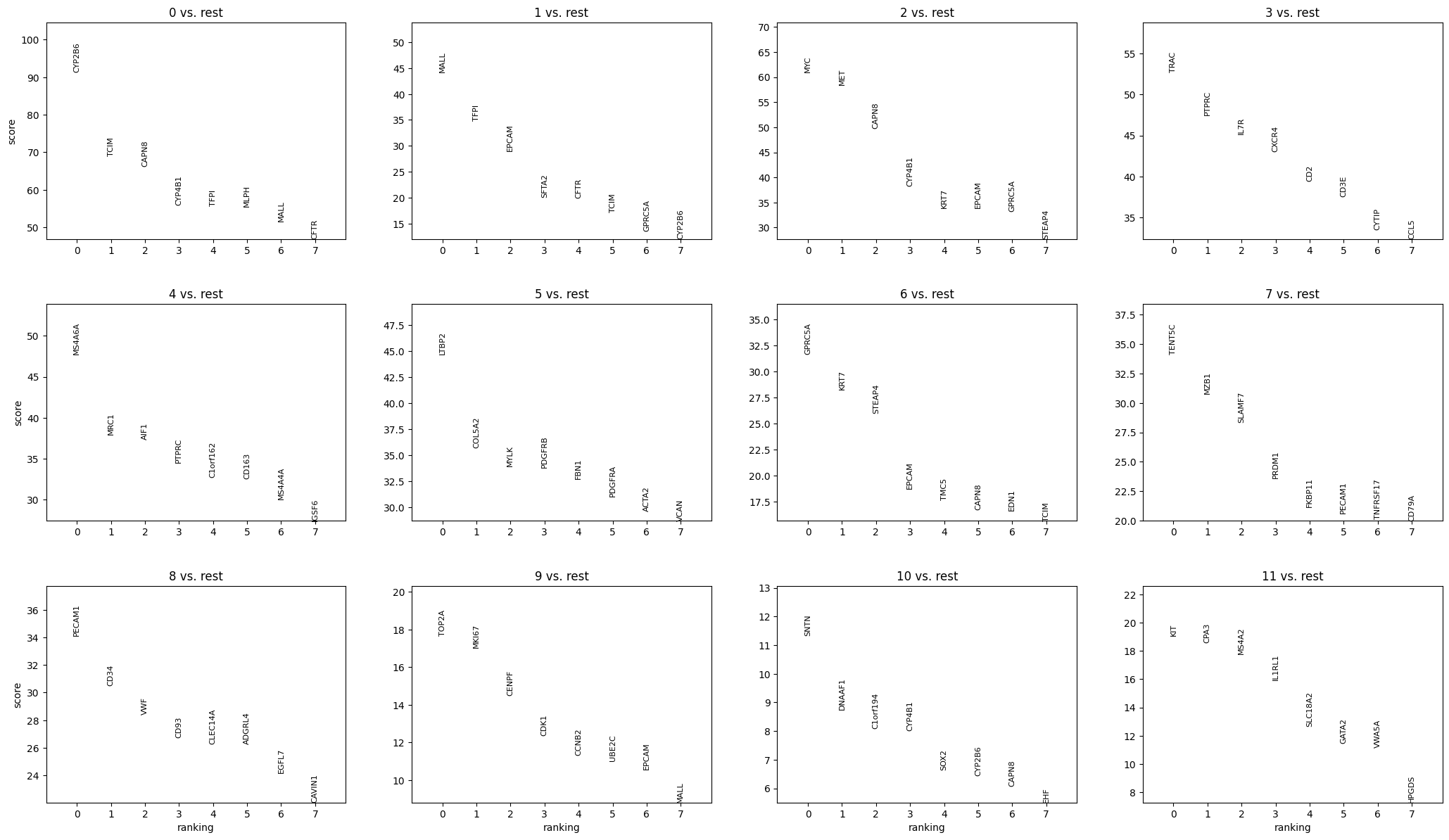
sp.pl.cluster(
sdata,
table_layer="table_dapi_preprocessed",
key_added="leiden",
)
sp.pl.plot_shapes(
sdata,
img_layer="DAPI",
linewidth=0.3,
table_layer="table_dapi_preprocessed",
column="leiden",
shapes_layer="segmentation_mask_boundaries_dapi",
)

Cell segmentation: hierarchical approach#
For the second approach, we additionally segment also the boundary and interior RNA stain. We will adapt a hierarchical approach, inspired by the segmentation method applied by 10X for the Xenium analysis pipeline, imaged below. First priority is given to the boundary stain, all cells segmented from this stain will be added to the final segmentation mask. Next priority is given to the interior RNA stain, all cells segmented in the interior RNA stain, that have not been segmented already in the boundary stain, and that have an overlapping segmented nucleus from the DAPI stain, will be added to the final segmentation mask. Finally, if any nuclei have been segmented in the DAPI stain that do not overlap with a cell from the boundary or interior RNA stain, these will also be included in the final segmentation mask.

# Similar to what we did with the DAPI stain, we will separate out the interior RNA and boundary stain in order to segment them
sdata["interior_RNA"] = sdata["clahe"][1:2, :, :]
sdata["boundary"] = sdata["clahe"][2:3, :, :]
sdata
SpatialData object, with associated Zarr store: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr
├── Images
│ ├── 'DAPI': DataArray[cyx] (1, 6000, 7000)
│ ├── 'boundary': DataArray[cyx] (1, 6000, 7000)
│ ├── 'clahe': DataArray[cyx] (4, 6000, 7000)
│ ├── 'he_image_global': DataTree[cyx] (3, 45087, 11580), (3, 22543, 5790), (3, 11271, 2895), (3, 5635, 1447), (3, 2817, 723)
│ ├── 'interior_RNA': DataArray[cyx] (1, 6000, 7000)
│ ├── 'min_max_filtered': DataArray[cyx] (4, 6000, 7000)
│ └── 'morphology_focus_global': DataTree[cyx] (4, 17098, 51187), (4, 8549, 25593), (4, 4274, 12796), (4, 2137, 6398), (4, 1068, 3199)
├── Labels
│ ├── 'cell_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
│ ├── 'nucleus_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
│ └── 'segmentation_mask_dapi': DataArray[yx] (6000, 7000)
├── Points
│ └── 'transcripts_global': DataFrame with shape: (<Delayed>, 3) (2D points)
├── Shapes
│ ├── 'filtered_low_counts_segmentation_mask_boundaries_dapi': GeoDataFrame shape: (711, 1) (2D shapes)
│ ├── 'filtered_size_segmentation_mask_boundaries_dapi': GeoDataFrame shape: (12, 1) (2D shapes)
│ └── 'segmentation_mask_boundaries_dapi': GeoDataFrame shape: (19227, 1) (2D shapes)
└── Tables
├── 'table_dapi': AnnData (19894, 377)
└── 'table_dapi_preprocessed': AnnData (19227, 377)
with coordinate systems:
▸ 'global', with elements:
DAPI (Images), boundary (Images), clahe (Images), he_image_global (Images), interior_RNA (Images), min_max_filtered (Images), morphology_focus_global (Images), cell_labels_global (Labels), nucleus_labels_global (Labels), segmentation_mask_dapi (Labels), transcripts_global (Points), filtered_low_counts_segmentation_mask_boundaries_dapi (Shapes), filtered_size_segmentation_mask_boundaries_dapi (Shapes), segmentation_mask_boundaries_dapi (Shapes)
with the following Dask-backed elements not being self-contained:
▸ DAPI: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
▸ interior_RNA: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
▸ boundary: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
with the following elements not in the Zarr store:
▸ interior_RNA (Images)
▸ boundary (Images)
▸ DAPI (Images)
crd=[21593, 28593, 6549, 12549]
sdata = sp.im.segment(sdata=sdata,
img_layer='interior_RNA',
output_labels_layer='segmentation_mask_interior_RNA',
output_shapes_layer='segmentation_mask_boundaries_interior_RNA',
crd=crd,
to_coordinate_system='global',
flow_threshold=0.95,
diameter=55,
cellprob_threshold=-6,
min_size=30,
pretrained_model='cyto3',
overwrite=True)
sp.pl.segment(sdata=sdata, crd=crd, img_layer="interior_RNA", shapes_layer="segmentation_mask_boundaries_interior_RNA")

crd=[21593, 28593, 6549, 12549]
sdata = sp.im.segment(sdata=sdata,
img_layer='boundary',
output_labels_layer='segmentation_mask_boundary',
output_shapes_layer='segmentation_mask_boundaries_boundary',
crd=crd,
to_coordinate_system='global',
flow_threshold=0.90,
diameter=45,
cellprob_threshold=-4,
min_size=30,
pretrained_model='cyto3',
overwrite=True)
sp.pl.segment(sdata=sdata, crd=crd, img_layer="boundary", shapes_layer="segmentation_mask_boundaries_boundary")

# First, we will filter out any cells segmented from the interior RNA stain that do not overlap with segmented nuclei from the DAPI stain
sdata = sp.im.align_labels_layers(sdata,
labels_layer_1='segmentation_mask_interior_RNA',
labels_layer_2='segmentation_mask_dapi',
threshold=0.3,
chunks=(2000, 2000),
output_labels_layer='segmentation_mask_interior_RNA_overlap_dapi',
output_shapes_layer='segmentation_mask_boundaries_interior_RNA_overlap_dapi',
scale_factors=None,
overwrite=True)
sdata
SpatialData object, with associated Zarr store: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr
├── Images
│ ├── 'DAPI': DataArray[cyx] (1, 6000, 7000)
│ ├── 'boundary': DataArray[cyx] (1, 6000, 7000)
│ ├── 'clahe': DataArray[cyx] (4, 6000, 7000)
│ ├── 'he_image_global': DataTree[cyx] (3, 45087, 11580), (3, 22543, 5790), (3, 11271, 2895), (3, 5635, 1447), (3, 2817, 723)
│ ├── 'interior_RNA': DataArray[cyx] (1, 6000, 7000)
│ ├── 'min_max_filtered': DataArray[cyx] (4, 6000, 7000)
│ └── 'morphology_focus_global': DataTree[cyx] (4, 17098, 51187), (4, 8549, 25593), (4, 4274, 12796), (4, 2137, 6398), (4, 1068, 3199)
├── Labels
│ ├── 'cell_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
│ ├── 'nucleus_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
│ ├── 'segmentation_mask_boundary': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_dapi': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_interior_RNA': DataArray[yx] (6000, 7000)
│ └── 'segmentation_mask_interior_RNA_overlap_dapi': DataArray[yx] (6000, 7000)
├── Points
│ └── 'transcripts_global': DataFrame with shape: (<Delayed>, 3) (2D points)
├── Shapes
│ ├── 'filtered_low_counts_segmentation_mask_boundaries_dapi': GeoDataFrame shape: (711, 1) (2D shapes)
│ ├── 'filtered_size_segmentation_mask_boundaries_dapi': GeoDataFrame shape: (12, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_boundary': GeoDataFrame shape: (15906, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_dapi': GeoDataFrame shape: (19227, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_interior_RNA': GeoDataFrame shape: (16056, 1) (2D shapes)
│ └── 'segmentation_mask_boundaries_interior_RNA_overlap_dapi': GeoDataFrame shape: (11659, 1) (2D shapes)
└── Tables
├── 'table_dapi': AnnData (19894, 377)
└── 'table_dapi_preprocessed': AnnData (19227, 377)
with coordinate systems:
▸ 'global', with elements:
DAPI (Images), boundary (Images), clahe (Images), he_image_global (Images), interior_RNA (Images), min_max_filtered (Images), morphology_focus_global (Images), cell_labels_global (Labels), nucleus_labels_global (Labels), segmentation_mask_boundary (Labels), segmentation_mask_dapi (Labels), segmentation_mask_interior_RNA (Labels), segmentation_mask_interior_RNA_overlap_dapi (Labels), transcripts_global (Points), filtered_low_counts_segmentation_mask_boundaries_dapi (Shapes), filtered_size_segmentation_mask_boundaries_dapi (Shapes), segmentation_mask_boundaries_boundary (Shapes), segmentation_mask_boundaries_dapi (Shapes), segmentation_mask_boundaries_interior_RNA (Shapes), segmentation_mask_boundaries_interior_RNA_overlap_dapi (Shapes)
with the following Dask-backed elements not being self-contained:
▸ DAPI: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
▸ interior_RNA: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
▸ boundary: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
with the following elements not in the Zarr store:
▸ interior_RNA (Images)
▸ boundary (Images)
▸ DAPI (Images)
# Next, we merge these filtered cells, with the cells segmented from the boundary stain, since segmentation_mask_boundary is layer 1 here, all these cells will be kept, giving it first priority
sp.im.merge_labels_layers(sdata,
labels_layer_1='segmentation_mask_boundary',
labels_layer_2='segmentation_mask_interior_RNA_overlap_dapi',
threshold=0.5,
chunks=(2000, 2000),
output_labels_layer='segmentation_mask_merge_1',
output_shapes_layer='segmentation_mask_boundaries_merge_1',
overwrite=True)
SpatialData object, with associated Zarr store: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr
├── Images
│ ├── 'DAPI': DataArray[cyx] (1, 6000, 7000)
│ ├── 'boundary': DataArray[cyx] (1, 6000, 7000)
│ ├── 'clahe': DataArray[cyx] (4, 6000, 7000)
│ ├── 'he_image_global': DataTree[cyx] (3, 45087, 11580), (3, 22543, 5790), (3, 11271, 2895), (3, 5635, 1447), (3, 2817, 723)
│ ├── 'interior_RNA': DataArray[cyx] (1, 6000, 7000)
│ ├── 'min_max_filtered': DataArray[cyx] (4, 6000, 7000)
│ └── 'morphology_focus_global': DataTree[cyx] (4, 17098, 51187), (4, 8549, 25593), (4, 4274, 12796), (4, 2137, 6398), (4, 1068, 3199)
├── Labels
│ ├── 'cell_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
│ ├── 'nucleus_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
│ ├── 'segmentation_mask_boundary': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_dapi': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_interior_RNA': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_interior_RNA_overlap_dapi': DataArray[yx] (6000, 7000)
│ └── 'segmentation_mask_merge_1': DataArray[yx] (6000, 7000)
├── Points
│ └── 'transcripts_global': DataFrame with shape: (<Delayed>, 3) (2D points)
├── Shapes
│ ├── 'filtered_low_counts_segmentation_mask_boundaries_dapi': GeoDataFrame shape: (711, 1) (2D shapes)
│ ├── 'filtered_size_segmentation_mask_boundaries_dapi': GeoDataFrame shape: (12, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_boundary': GeoDataFrame shape: (15906, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_dapi': GeoDataFrame shape: (19227, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_interior_RNA': GeoDataFrame shape: (16056, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_interior_RNA_overlap_dapi': GeoDataFrame shape: (11659, 1) (2D shapes)
│ └── 'segmentation_mask_boundaries_merge_1': GeoDataFrame shape: (17960, 1) (2D shapes)
└── Tables
├── 'table_dapi': AnnData (19894, 377)
└── 'table_dapi_preprocessed': AnnData (19227, 377)
with coordinate systems:
▸ 'global', with elements:
DAPI (Images), boundary (Images), clahe (Images), he_image_global (Images), interior_RNA (Images), min_max_filtered (Images), morphology_focus_global (Images), cell_labels_global (Labels), nucleus_labels_global (Labels), segmentation_mask_boundary (Labels), segmentation_mask_dapi (Labels), segmentation_mask_interior_RNA (Labels), segmentation_mask_interior_RNA_overlap_dapi (Labels), segmentation_mask_merge_1 (Labels), transcripts_global (Points), filtered_low_counts_segmentation_mask_boundaries_dapi (Shapes), filtered_size_segmentation_mask_boundaries_dapi (Shapes), segmentation_mask_boundaries_boundary (Shapes), segmentation_mask_boundaries_dapi (Shapes), segmentation_mask_boundaries_interior_RNA (Shapes), segmentation_mask_boundaries_interior_RNA_overlap_dapi (Shapes), segmentation_mask_boundaries_merge_1 (Shapes)
with the following Dask-backed elements not being self-contained:
▸ DAPI: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
▸ interior_RNA: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
▸ boundary: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
with the following elements not in the Zarr store:
▸ interior_RNA (Images)
▸ boundary (Images)
▸ DAPI (Images)
# We do the same but now with the dapi segmentation mask, to get the final segmentation mask
sp.im.merge_labels_layers(sdata,
labels_layer_1='segmentation_mask_merge_1',
labels_layer_2='segmentation_mask_dapi',
threshold=0.5,
chunks=(2000, 2000),
output_labels_layer='segmentation_mask_merge_2',
output_shapes_layer='segmentation_mask_boundaries_merge_2',
overwrite=True)
SpatialData object, with associated Zarr store: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr
├── Images
│ ├── 'DAPI': DataArray[cyx] (1, 6000, 7000)
│ ├── 'boundary': DataArray[cyx] (1, 6000, 7000)
│ ├── 'clahe': DataArray[cyx] (4, 6000, 7000)
│ ├── 'he_image_global': DataTree[cyx] (3, 45087, 11580), (3, 22543, 5790), (3, 11271, 2895), (3, 5635, 1447), (3, 2817, 723)
│ ├── 'interior_RNA': DataArray[cyx] (1, 6000, 7000)
│ ├── 'min_max_filtered': DataArray[cyx] (4, 6000, 7000)
│ └── 'morphology_focus_global': DataTree[cyx] (4, 17098, 51187), (4, 8549, 25593), (4, 4274, 12796), (4, 2137, 6398), (4, 1068, 3199)
├── Labels
│ ├── 'cell_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
│ ├── 'nucleus_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
│ ├── 'segmentation_mask_boundary': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_dapi': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_interior_RNA': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_interior_RNA_overlap_dapi': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_merge_1': DataArray[yx] (6000, 7000)
│ └── 'segmentation_mask_merge_2': DataArray[yx] (6000, 7000)
├── Points
│ └── 'transcripts_global': DataFrame with shape: (<Delayed>, 3) (2D points)
├── Shapes
│ ├── 'filtered_low_counts_segmentation_mask_boundaries_dapi': GeoDataFrame shape: (711, 1) (2D shapes)
│ ├── 'filtered_size_segmentation_mask_boundaries_dapi': GeoDataFrame shape: (12, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_boundary': GeoDataFrame shape: (15906, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_dapi': GeoDataFrame shape: (19227, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_interior_RNA': GeoDataFrame shape: (16056, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_interior_RNA_overlap_dapi': GeoDataFrame shape: (11659, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_merge_1': GeoDataFrame shape: (17960, 1) (2D shapes)
│ └── 'segmentation_mask_boundaries_merge_2': GeoDataFrame shape: (20516, 1) (2D shapes)
└── Tables
├── 'table_dapi': AnnData (19894, 377)
└── 'table_dapi_preprocessed': AnnData (19227, 377)
with coordinate systems:
▸ 'global', with elements:
DAPI (Images), boundary (Images), clahe (Images), he_image_global (Images), interior_RNA (Images), min_max_filtered (Images), morphology_focus_global (Images), cell_labels_global (Labels), nucleus_labels_global (Labels), segmentation_mask_boundary (Labels), segmentation_mask_dapi (Labels), segmentation_mask_interior_RNA (Labels), segmentation_mask_interior_RNA_overlap_dapi (Labels), segmentation_mask_merge_1 (Labels), segmentation_mask_merge_2 (Labels), transcripts_global (Points), filtered_low_counts_segmentation_mask_boundaries_dapi (Shapes), filtered_size_segmentation_mask_boundaries_dapi (Shapes), segmentation_mask_boundaries_boundary (Shapes), segmentation_mask_boundaries_dapi (Shapes), segmentation_mask_boundaries_interior_RNA (Shapes), segmentation_mask_boundaries_interior_RNA_overlap_dapi (Shapes), segmentation_mask_boundaries_merge_1 (Shapes), segmentation_mask_boundaries_merge_2 (Shapes)
with the following Dask-backed elements not being self-contained:
▸ DAPI: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
▸ interior_RNA: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
▸ boundary: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
with the following elements not in the Zarr store:
▸ interior_RNA (Images)
▸ boundary (Images)
▸ DAPI (Images)
# We cam plot this final mask on all four channels
crd=[21593, 28593, 6549, 12549]
sp.pl.segment(sdata=sdata, crd=crd, img_layer="clahe", shapes_layer="segmentation_mask_boundaries_merge_2", channel=['DAPI', '18S', 'ATP1A1/CD45/E-Cadherin'])

# Allocation of the transcripts
sdata = sp.tb.allocate(
sdata=sdata,
labels_layer="segmentation_mask_merge_2",
points_layer="transcripts_global",
output_layer="table_merge",
update_shapes_layers=False,
overwrite=True,
)
sdata['table_merge']
AnnData object with n_obs × n_vars = 20434 × 377
obs: 'cell_ID', 'fov_labels'
uns: 'spatialdata_attrs'
obsm: 'spatial'
# Perform preprocessing.
sdata = sp.tb.preprocess_transcriptomics(
sdata,
labels_layer="segmentation_mask_merge_2",
table_layer="table_merge",
output_layer="table_merge_preprocessed",
min_counts=10,
min_cells=5,
size_norm=True,
n_comps=50,
overwrite=True,
update_shapes_layers=True,
shapes_layers_to_filter=['segmentation_mask_boundaries_merge_2']
)
# NOTE: We're skipping a bunch of QC-steps here. For more details about this, see general tutorial
# Now we can plot cell sizes
sp.pl.plot_shapes(
sdata,
img_layer="boundary",
table_layer="table_merge_preprocessed",
column="shapeSize",
crd=crd,
shapes_layer="segmentation_mask_boundaries_merge_2",
linewidth=0.2,
alpha=0.7
)
# We can further filter on cell size in order to discard any noise or segmentation mistakes
sdata = sp.tb.filter_on_size(
sdata,
labels_layer="segmentation_mask_merge_2",
table_layer="table_merge_preprocessed",
output_layer="table_merge_preprocessed",
min_size=500,
max_size=10000,
overwrite=True,
shapes_layers_to_filter=['segmentation_mask_boundaries_merge_2']
)
sdata = sp.tb.leiden(
sdata,
labels_layer="segmentation_mask_merge_2",
table_layer="table_merge_preprocessed",
output_layer="table_merge_preprocessed",
calculate_umap=True,
calculate_neighbors=True,
n_pcs=17,
n_neighbors=35,
resolution=0.8,
rank_genes=True,
key_added="leiden",
overwrite=True,
)
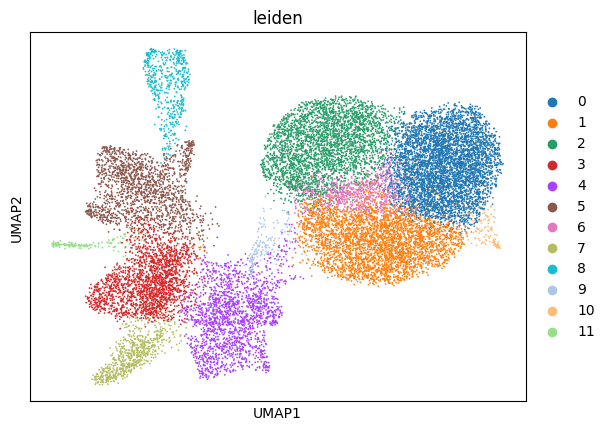
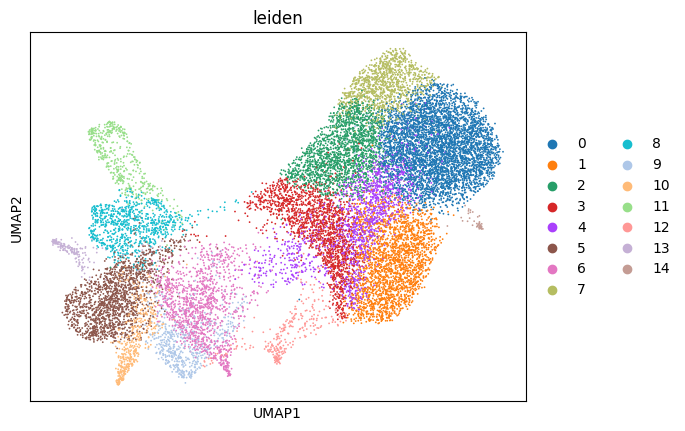
sp.pl.cluster(
sdata,
table_layer="table_merge_preprocessed",
key_added="leiden",
)
sp.pl.plot_shapes(
sdata,
img_layer="boundary",
linewidth=0.3,
alpha=0.5,
table_layer="table_merge_preprocessed",
column="leiden",
shapes_layer="segmentation_mask_boundaries_merge_2",
)

Cell segmentation: combined Cellpose#
Another approach to segment the multi-modal image, is to use the Cellpose segmentation model. It supports the input of a nuclei channel and a boundary channel.
from sparrow.image._image import _get_spatial_element
# Image channels: array(['DAPI', 'ATP1A1/CD45/E-Cadherin', '18S', 'AlphaSMA/Vimentin'],
se = _get_spatial_element(sdata, layer="clahe")
channels = [ se.c.data.tolist().index("ATP1A1/CD45/E-Cadherin" ) + 1,
se.c.data.tolist().index("DAPI" ) + 1 ] # + 1 because CellPose channel specifications are 1-indexed!!
channels # [membrane stain index + 1, nuclear stain index + 1]
[3, 1]
crd=[21593, 28593, 6549, 12549]
sdata = sp.im.segment(
sdata,
img_layer="clahe", # or "morphology_focus_global"
chunks=2048,
depth=200,
pretrained_model = "cyto3",
diameter=50,
flow_threshold=0.9,
cellprob_threshold=-4,
channels=channels,
output_labels_layer="segmentation_mask",
output_shapes_layer="segmentation_mask_boundaries",
crd=crd, # region to segment [x_min, xmax, y_min, y_max],
overwrite=True,
)
crd=[21593, 28593, 6549, 12549]
sp.pl.segment(sdata=sdata, crd=crd, img_layer="clahe", shapes_layer="segmentation_mask_boundaries", channel=['DAPI', '18S', 'ATP1A1/CD45/E-Cadherin'])

sdata = sp.tb.allocate(
sdata=sdata,
labels_layer="segmentation_mask", # we use the segmentation mask from Xenium, but we could also use CellPose segmentation masks in the layer "segmentation_mask"
points_layer="transcripts_global",
output_layer="table",
update_shapes_layers=False,
overwrite=True,
)
sdata['table']
AnnData object with n_obs × n_vars = 20769 × 377
obs: 'cell_ID', 'fov_labels'
uns: 'spatialdata_attrs'
obsm: 'spatial'
sdata
SpatialData object, with associated Zarr store: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr
├── Images
│ ├── 'DAPI': DataArray[cyx] (1, 6000, 7000)
│ ├── 'boundary': DataArray[cyx] (1, 6000, 7000)
│ ├── 'clahe': DataArray[cyx] (4, 6000, 7000)
│ ├── 'he_image_global': DataTree[cyx] (3, 45087, 11580), (3, 22543, 5790), (3, 11271, 2895), (3, 5635, 1447), (3, 2817, 723)
│ ├── 'interior_RNA': DataArray[cyx] (1, 6000, 7000)
│ ├── 'min_max_filtered': DataArray[cyx] (4, 6000, 7000)
│ └── 'morphology_focus_global': DataTree[cyx] (4, 17098, 51187), (4, 8549, 25593), (4, 4274, 12796), (4, 2137, 6398), (4, 1068, 3199)
├── Labels
│ ├── 'cell_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
│ ├── 'nucleus_labels_global': DataTree[yx] (17098, 51187), (8549, 25593), (4274, 12796), (2137, 6398), (1068, 3199)
│ ├── 'segmentation_mask': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_boundary': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_dapi': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_interior_RNA': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_interior_RNA_overlap_dapi': DataArray[yx] (6000, 7000)
│ ├── 'segmentation_mask_merge_1': DataArray[yx] (6000, 7000)
│ └── 'segmentation_mask_merge_2': DataArray[yx] (6000, 7000)
├── Points
│ └── 'transcripts_global': DataFrame with shape: (<Delayed>, 3) (2D points)
├── Shapes
│ ├── 'filtered_low_counts_segmentation_mask_boundaries_dapi': GeoDataFrame shape: (711, 1) (2D shapes)
│ ├── 'filtered_low_counts_segmentation_mask_boundaries_merge_2': GeoDataFrame shape: (913, 1) (2D shapes)
│ ├── 'filtered_size_segmentation_mask_boundaries_dapi': GeoDataFrame shape: (12, 1) (2D shapes)
│ ├── 'filtered_size_segmentation_mask_boundaries_merge_2': GeoDataFrame shape: (1377, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries': GeoDataFrame shape: (20895, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_boundary': GeoDataFrame shape: (15906, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_dapi': GeoDataFrame shape: (19227, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_interior_RNA': GeoDataFrame shape: (16056, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_interior_RNA_overlap_dapi': GeoDataFrame shape: (11659, 1) (2D shapes)
│ ├── 'segmentation_mask_boundaries_merge_1': GeoDataFrame shape: (17960, 1) (2D shapes)
│ └── 'segmentation_mask_boundaries_merge_2': GeoDataFrame shape: (18226, 1) (2D shapes)
└── Tables
├── 'table': AnnData (20769, 377)
├── 'table_dapi': AnnData (19894, 377)
├── 'table_dapi_preprocessed': AnnData (19227, 377)
├── 'table_merge': AnnData (20434, 377)
└── 'table_merge_preprocessed': AnnData (18226, 377)
with coordinate systems:
▸ 'global', with elements:
DAPI (Images), boundary (Images), clahe (Images), he_image_global (Images), interior_RNA (Images), min_max_filtered (Images), morphology_focus_global (Images), cell_labels_global (Labels), nucleus_labels_global (Labels), segmentation_mask (Labels), segmentation_mask_boundary (Labels), segmentation_mask_dapi (Labels), segmentation_mask_interior_RNA (Labels), segmentation_mask_interior_RNA_overlap_dapi (Labels), segmentation_mask_merge_1 (Labels), segmentation_mask_merge_2 (Labels), transcripts_global (Points), filtered_low_counts_segmentation_mask_boundaries_dapi (Shapes), filtered_low_counts_segmentation_mask_boundaries_merge_2 (Shapes), filtered_size_segmentation_mask_boundaries_dapi (Shapes), filtered_size_segmentation_mask_boundaries_merge_2 (Shapes), segmentation_mask_boundaries (Shapes), segmentation_mask_boundaries_boundary (Shapes), segmentation_mask_boundaries_dapi (Shapes), segmentation_mask_boundaries_interior_RNA (Shapes), segmentation_mask_boundaries_interior_RNA_overlap_dapi (Shapes), segmentation_mask_boundaries_merge_1 (Shapes), segmentation_mask_boundaries_merge_2 (Shapes)
with the following Dask-backed elements not being self-contained:
▸ DAPI: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
▸ interior_RNA: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
▸ boundary: /srv/scratch/karenh/sparrow-revision/data/sdata_objects/sdata_Xenium_lung_multistaining_tutorial.zarr/images/clahe
with the following elements not in the Zarr store:
▸ interior_RNA (Images)
▸ boundary (Images)
▸ DAPI (Images)
# Perform preprocessing.
sdata = sp.tb.preprocess_transcriptomics(
sdata,
labels_layer="segmentation_mask",
table_layer="table",
output_layer="table_preprocessed",
min_counts=10,
min_cells=5,
size_norm=True,
n_comps=50,
overwrite=True,
update_shapes_layers=True,
shapes_layers_to_filter=['segmentation_mask_boundaries']
)
# NOTE: We're skipping a bunch of QC-steps here. For more details about this, see general tutorial
sp.pl.plot_shapes(
sdata,
img_layer="clahe",
table_layer="table_preprocessed",
column="shapeSize",
crd=crd,
shapes_layer="segmentation_mask_boundaries",
linewidth=0.2,
alpha=0.7,
)
sdata = sp.tb.filter_on_size(
sdata,
labels_layer="segmentation_mask",
table_layer="table_preprocessed",
output_layer="table_preprocessed",
min_size=500,
max_size=8000,
overwrite=True,
shapes_layers_to_filter=['segmentation_mask_boundaries']
)
sdata = sp.tb.leiden(
sdata,
labels_layer="segmentation_mask",
table_layer="table_preprocessed",
output_layer="table_preprocessed",
calculate_umap=True,
calculate_neighbors=True,
n_pcs=17,
n_neighbors=35,
resolution=0.8,
rank_genes=True,
key_added="leiden",
overwrite=True,
)
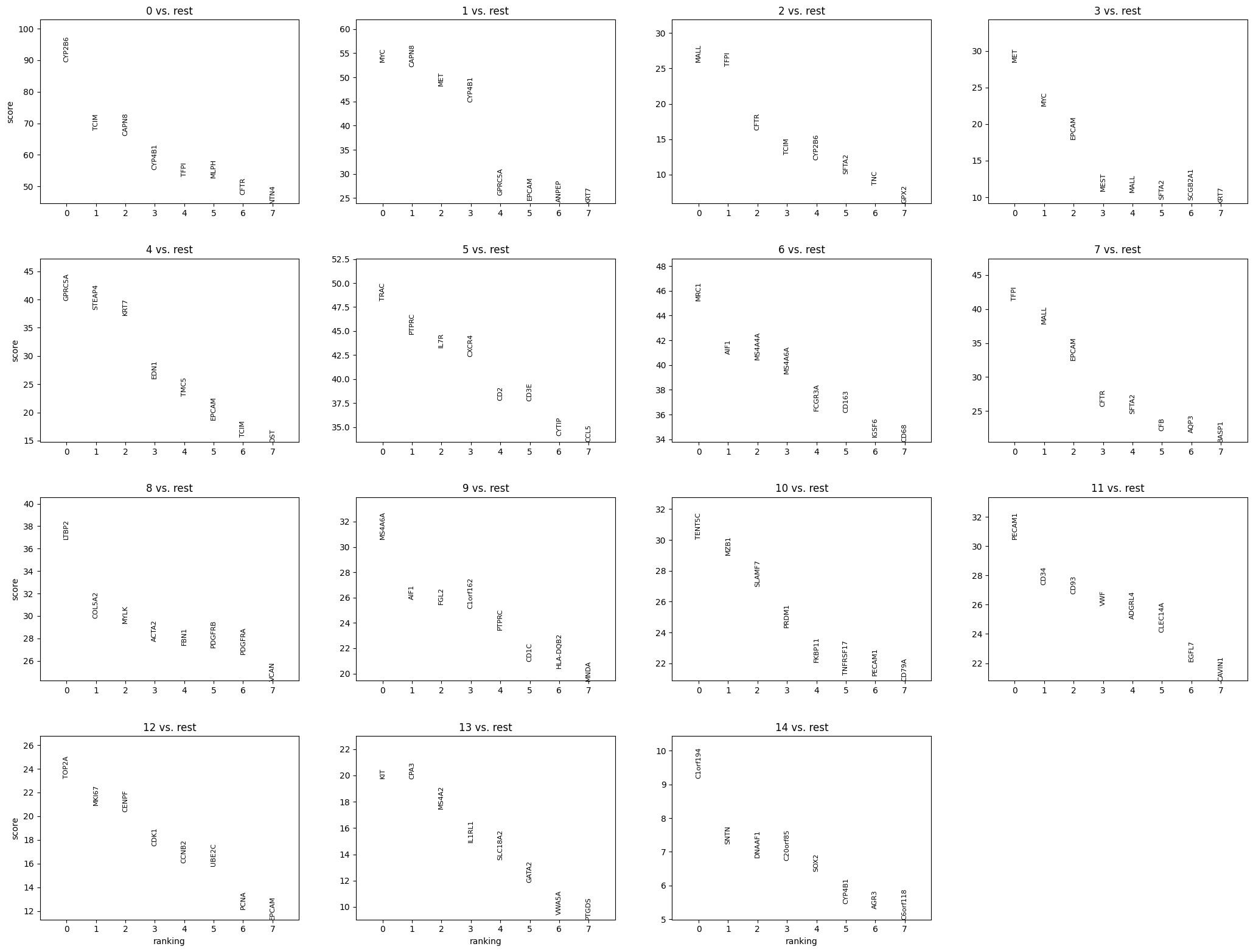
sp.pl.cluster(
sdata,
table_layer="table_preprocessed",
key_added="leiden",
)
sp.pl.plot_shapes(
sdata,
img_layer="boundary",
linewidth=0.3,
alpha=0.5,
table_layer="table_preprocessed",
column="leiden",
shapes_layer="segmentation_mask_boundaries",
)