Analyzing MERFISH Data from the MERSCOPE Platform#
This tutorial illustrates how to analyze MERFISH spatial transcriptomics data using SPArrOW.
We will use a MERFISH mouse liver dataset generated on the Vizgen MERSCOPE platform, downloaded from https://info.vizgen.com/mouse-liver-data (accessed 2024-01-10).
Installation#
uv venv .venv_sparrow --python=3.12
source .venv_sparrow/bin/activate
uv pip install -e .
uv pip install jupyter
uv pip install rioxarray
uv pip install cellpose==3.1.1.1
uv pip install dask-cuda==24.12.0
uv pip install bokeh
%load_ext autoreload
%autoreload 2
import sparrow as sp
Download the data.#
from sparrow.datasets.registry import get_registry
registry = get_registry()
_ = registry.fetch("transcriptomics/vizgen/mouse/Liver1Slice1/images/mosaic_DAPI_z3.tif")
_ = registry.fetch("transcriptomics/vizgen/mouse/Liver1Slice1/images/mosaic_PolyT_z3.tif")
_ = registry.fetch("transcriptomics/vizgen/mouse/Liver1Slice1/images/micron_to_mosaic_pixel_transform.csv")
path_transcripts = registry.fetch("transcriptomics/vizgen/mouse/Liver1Slice1/detected_transcripts.csv")
Create the SpatialData object.#
import os
import tempfile
input_path = os.path.dirname(path_transcripts)
OUTPUT_DIR = tempfile.gettempdir()
# This step can take around 20 minutes.
sdata = sp.io.merscope(
path=input_path,
to_coordinate_system="global",
z_layers=[
3,
],
backend=None,
transcripts=True,
mosaic_images=True,
do_3D=False,
z_projection=False,
image_models_kwargs={"scale_factors": None},
output=os.path.join( OUTPUT_DIR, "sdata_merscope.zarr"),
filter_gene_names=[ "blank" ],
)
sp.pl.plot_shapes(
sdata,
img_layer=["mouse_Liver1Slice1_z3_global"],
crd = [ 50000, 60000, 40000, 50000 ],
channel="DAPI",
figsize = (5,5),
)
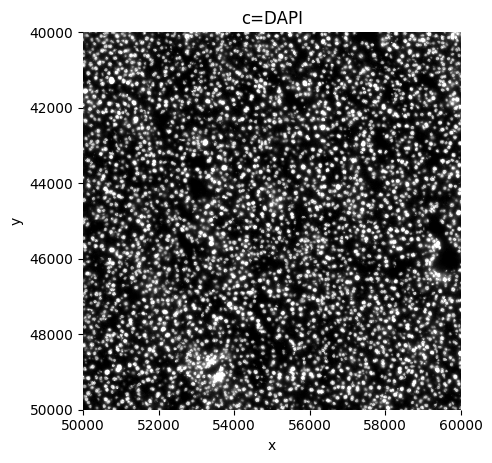
Preprocessing#
Note that we work on a crop for the rest of this tutorial.
sdata=sp.im.min_max_filtering(
sdata,
img_layer="mouse_Liver1Slice1_z3_global",
output_layer="min_max_filtered",
size_min_max_filter=[ 85, 135 ],
crd = [ 50000, 60000, 40000, 50000 ],
overwrite=True,
)
sdata=sp.im.enhance_contrast(
sdata,
img_layer="min_max_filtered",
output_layer="clahe",
contrast_clip=[13.5, 18.5 ],
crd = [ 50000, 60000, 40000, 50000 ],
overwrite=True,
)
sp.pl.plot_shapes(
sdata,
img_layer=["mouse_Liver1Slice1_z3_global", "clahe"],
crd = [ 50000, 60000, 40000, 50000 ],
figsize = (10,10),
)

Segmentation#
sdata[ "clahe" ].c.data
array(['DAPI', 'PolyT'], dtype='<U5')
# rechunk on disk
from spatialdata.transformations import get_transformation
sdata = sp.im.add_image_layer(
sdata,
arr =sdata[ "clahe" ].data.rechunk( 4096 ),
transformations=get_transformation( sdata[ "clahe"], get_all=True ),
output_layer = "clahe",
c_coords=sdata[ "clahe" ].c.data,
overwrite=True,
)
import torch
from dask.distributed import Client, LocalCluster
if torch.cuda.is_available():
from dask_cuda import LocalCUDACluster # pip install dask-cuda
# One worker per GPU; each worker will be pinned to a single GPU.
cluster = LocalCUDACluster(
CUDA_VISIBLE_DEVICES=[0], # or [0,1],...etc
n_workers=1, # 2 if [0,1],...etc
threads_per_worker=1,
memory_limit="32GB",
local_directory=os.environ.get( "TMPDIR" ),
)
else:
# cpu/mps fall back
from dask.distributed import LocalCluster
cluster = LocalCluster(
n_workers=1
if torch.backends.mps.is_available()
else 8, # If mps/cuda device available, it is better to increase chunk size to maximal value that fits on the gpu, and set n_workers to 1.
# For this dummy example, we only have one chunk, so setting n_workers>1, has no effect.
threads_per_worker=1,
memory_limit="32GB",
local_directory=os.environ.get( "TMPDIR" ),
)
client = Client(cluster)
print(client.dashboard_link)
http://127.0.0.1:36989/status
from sparrow.image._image import _get_spatial_element
se = _get_spatial_element( sdata, layer = "clahe" )
sdata = sp.im.segment(
sdata,
img_layer="clahe",
depth=200,
model=sp.im.cellpose_callable,
# parameters that will be passed to the callable sp.im.cellpose
pretrained_model = "cyto3",
diameter=100,
flow_threshold=0.85,
cellprob_threshold=-4,
channels = [ se.c.data.tolist().index("PolyT" )+1, se.c.data.tolist().index("DAPI" )+1 ],
output_labels_layer="segmentation_mask_crop",
output_shapes_layer="segmentation_mask_boundaries_crop",
crd= [50000, 60000, 40000, 50000], # region to segment [x_min, xmax, y_min, y_max],
overwrite=True,
)
client.close()
sp.pl.plot_shapes(
sdata,
shapes_layer="segmentation_mask_boundaries_crop",
img_layer=["clahe"],
crd = [ 50000, 55000, 40000, 45000 ],
figsize=( 10,10 ),
)

Create the AnnData table#
sdata = sp.tb.allocate(
sdata=sdata,
labels_layer="segmentation_mask_crop",
points_layer="transcripts_global",
output_layer="table_transcriptomics_crop",
update_shapes_layers=False,
overwrite=True,
)
sp.pl.sanity(
sdata,
img_layer="clahe",
shapes_layer = "segmentation_mask_boundaries_crop",
points_layer= "transcripts_global",
plot_cell_number=True,
gene="Vwf",
crd = [ 50500, 50500+500, 41000, 41500 ],
figsize=(5,5),
)
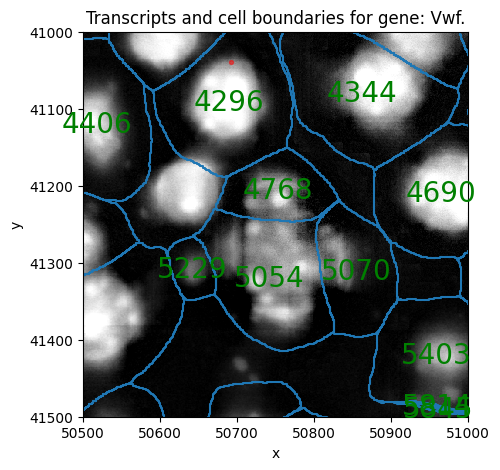
# Look-up a the number of transcripts for the Vwf gene in a cell shown above (using the cell ID),
# it should be the same number as transcripts plotted above.
sdata[ "table_transcriptomics_crop" ][sdata[ "table_transcriptomics_crop" ].obs[ "cell_ID" ] == 4296].to_df()["Vwf"]
cells
4296_segmentation_mask_crop_e0acaceb 1
Name: Vwf, dtype: uint32
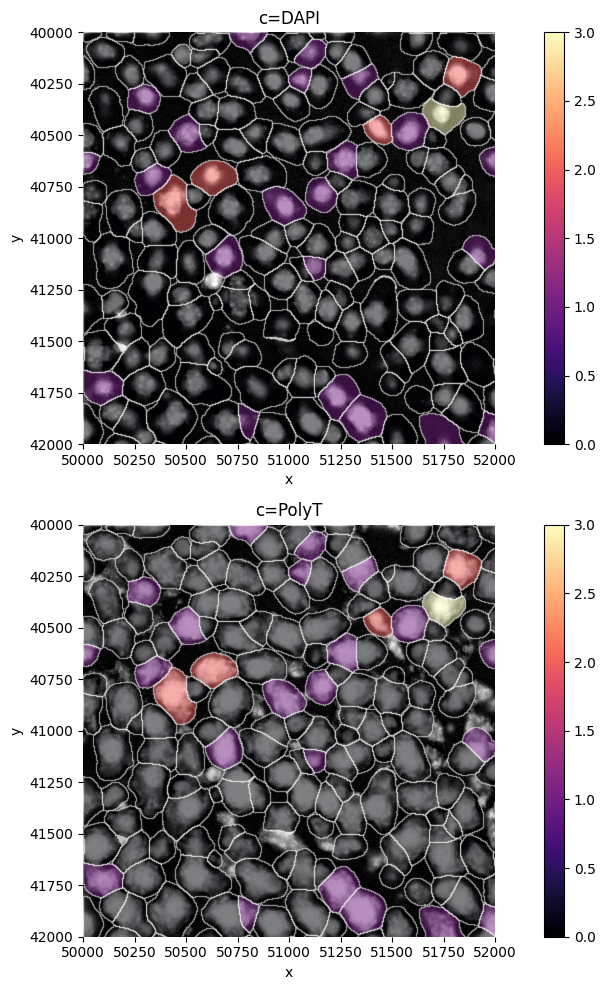
Visualize gene expression#
sp.pl.plot_shapes(
sdata,
img_layer="clahe",
shapes_layer="segmentation_mask_boundaries_crop",
figsize=( 10,10 ),
crd = [ 50000, 52000, 40000, 42000 ],
table_layer="table_transcriptomics_crop",
column = "Vwf",
)
df = sp.pl.analyse_genes_left_out(
sdata,
labels_layer="segmentation_mask_crop",
table_layer="table_transcriptomics_crop",
points_layer="transcripts_global",
)
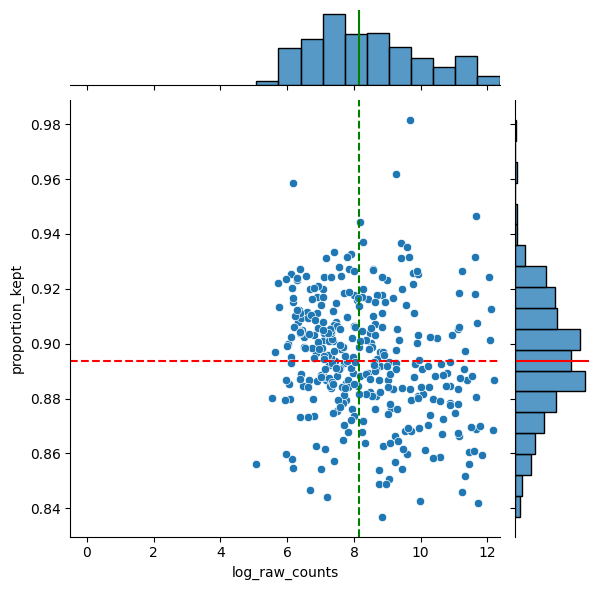
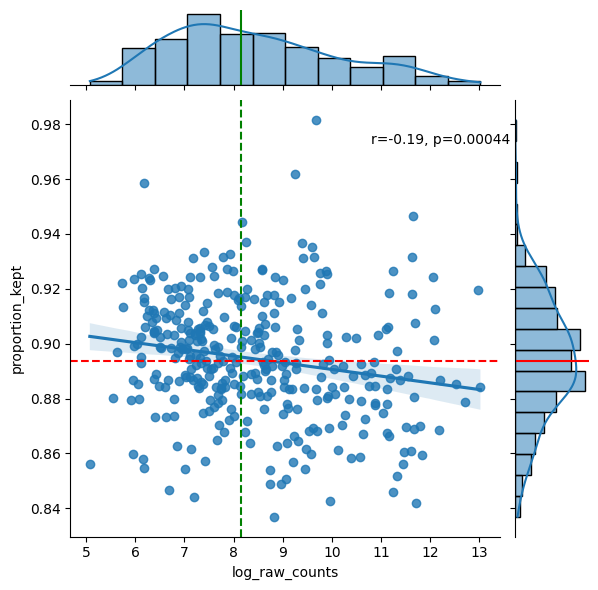
Preprocess the AnnData table#
# Perform preprocessing.
sdata = sp.tb.preprocess_transcriptomics(
sdata,
labels_layer="segmentation_mask_crop",
table_layer="table_transcriptomics_crop",
output_layer="table_transcriptomics_preprocessed_crop", # write results to a new slot, we could also write to the same slot (when passing overwrite==True).
min_counts=10,
min_cells=5,
size_norm=True,
n_comps=50,
overwrite=True,
update_shapes_layers=False,
)
sp.pl.preprocess_transcriptomics(
sdata,
table_layer="table_transcriptomics_preprocessed_crop",
)
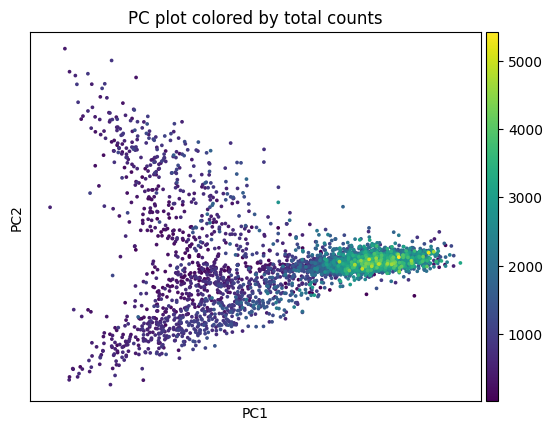
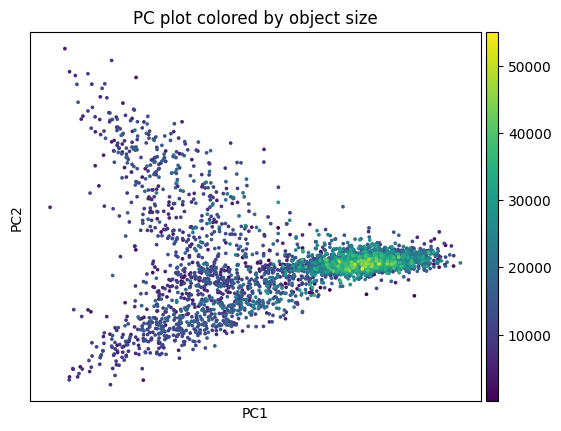
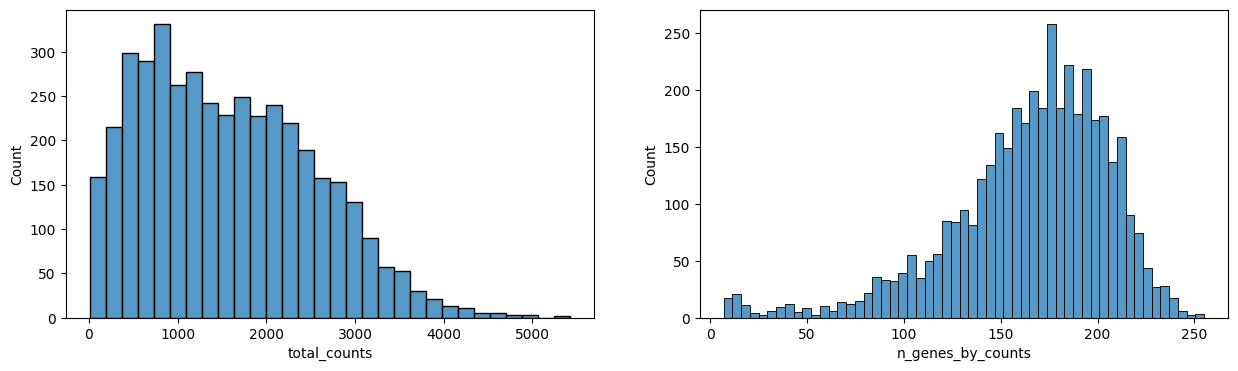
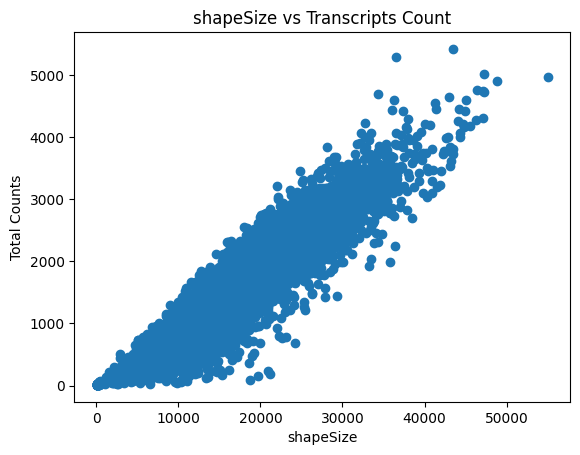
sdata = sp.tb.filter_on_size(
sdata,
labels_layer="segmentation_mask_crop",
table_layer="table_transcriptomics_preprocessed_crop",
output_layer="table_transcriptomics_filter_crop",
min_size=500,
max_size=100000,
update_shapes_layers=False,
overwrite=True,
)
Leiden clustering#
import scanpy as sc
sdata = sp.tb.leiden(
sdata,
labels_layer="segmentation_mask_crop",
table_layer="table_transcriptomics_filter_crop",
output_layer="table_transcriptomics_clustered_crop",
calculate_umap=True,
calculate_neighbors=True,
n_pcs=17,
n_neighbors=35,
resolution=1.0,
rank_genes=True,
key_added="leiden",
overwrite=True,
)
sc.pl.umap(sdata.tables["table_transcriptomics_clustered_crop"], color=["leiden"], show=True)
sc.pl.rank_genes_groups(sdata.tables["table_transcriptomics_clustered_crop"], n_genes=8, sharey=False, show=True)
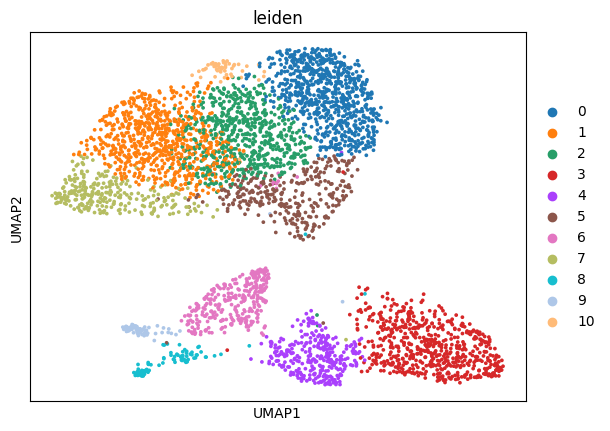
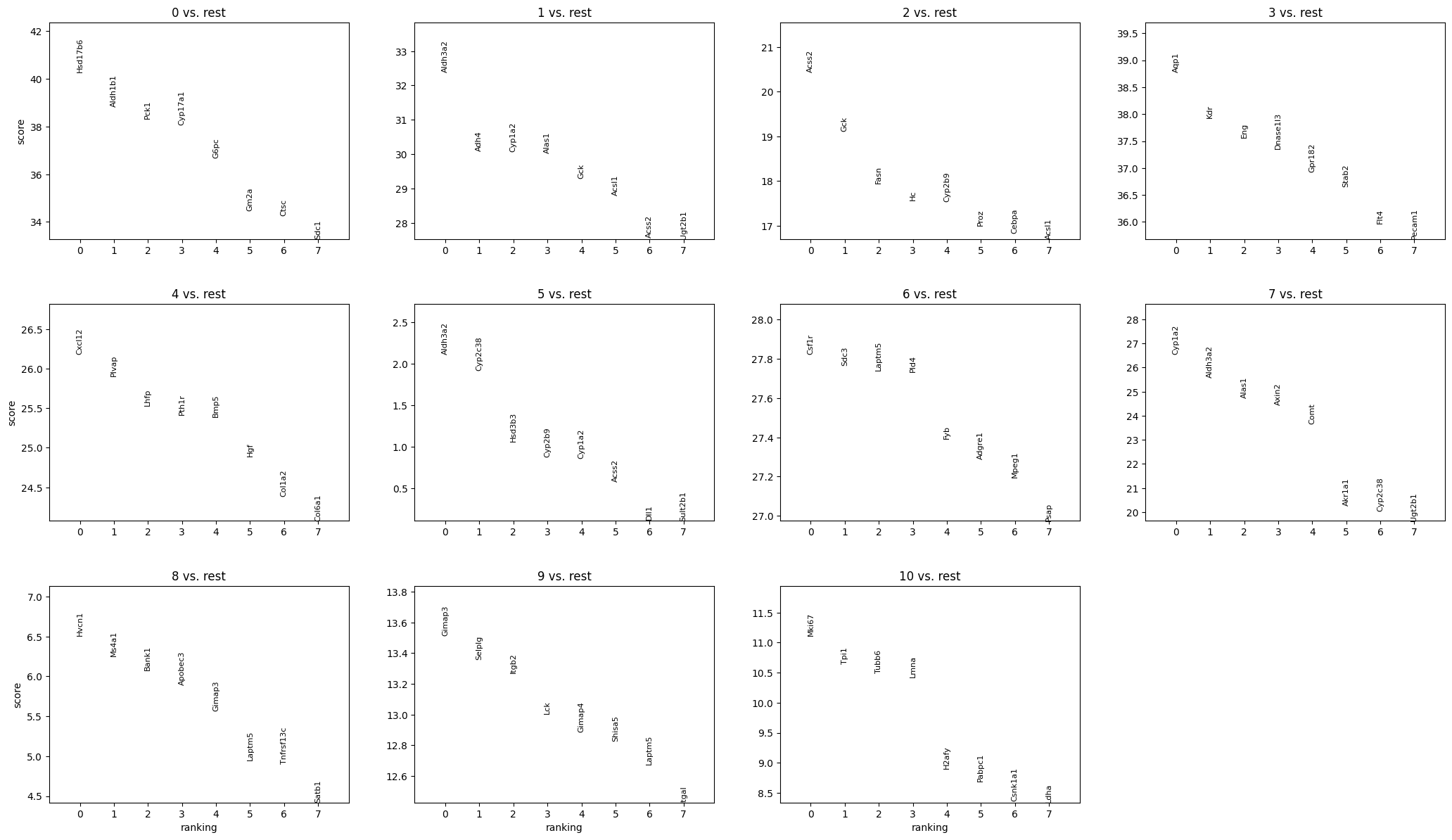
sdata[ "table_transcriptomics_clustered_crop" ].obs[ "leiden" ].head()
cells
149_segmentation_mask_crop_e0acaceb 7
181_segmentation_mask_crop_e0acaceb 7
229_segmentation_mask_crop_e0acaceb 1
264_segmentation_mask_crop_e0acaceb 1
293_segmentation_mask_crop_e0acaceb 3
Name: leiden, dtype: category
Categories (11, int64): [0, 1, 2, 3, ..., 7, 8, 9, 10]
sp.pl.plot_shapes(
sdata,
img_layer="clahe",
table_layer="table_transcriptomics_clustered_crop",
column="leiden",
shapes_layer="segmentation_mask_boundaries_crop",
alpha=1,
linewidth=0,
channel="DAPI",
crd = [ 50000, 60000, 40000, 50000 ],
)
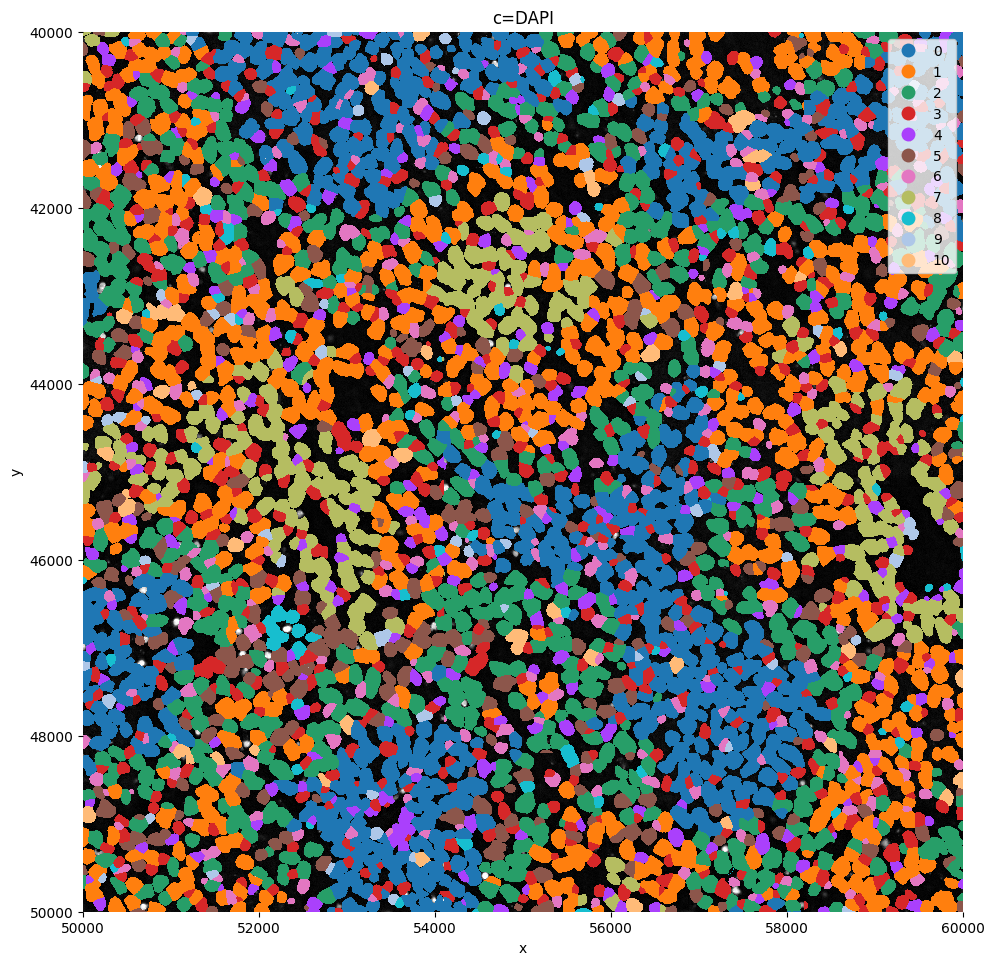
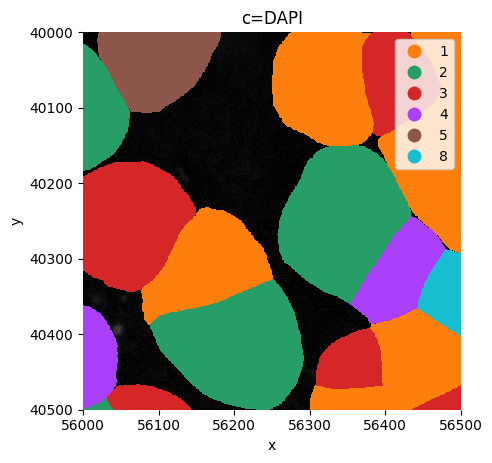
Or visualize a crop.
sp.pl.plot_shapes(
sdata,
img_layer="clahe",
table_layer="table_transcriptomics_clustered_crop",
column="leiden",
shapes_layer="segmentation_mask_boundaries_crop",
alpha=1,
linewidth=0,
channel="DAPI",
crd = [ 56000, 56000+500, 40000, 40500 ],
figsize=(5,5),
)